The talk of the world is coronavirus-so many questions. How did it come to infect humans? How should we go about controlling its spread? Will the virus mutate and become more infectious or deadly? Are there any effective treatments? There is no shortage of speculation and ongoing research hoping to provide answers to these questions. For example, as of April 14, 2020 there were 1630 manuscripts submitted to the preprint server for Biology https://www.biorxiv.org/ addressing all sorts of aspects of the new strain of coronavirus (SARS-CoV-2, COVID-19) in humans.
In addition to sharing my concerns about coronavirus back in February when the virus was sweeping through Europe, I have read a small sampling of those manuscripts and my students will each be reading two of them and then writing a paper on what they learned, so I will have 59 opportunities to learn a lot much more in the coming weeks.
I have been particularly interested in two COVID-related topics: the status of therapeutic treatments for COVID-19 and the evolutionary history of the broader coronavirus family. I want to tackle that second question today since I feel it has received the least amount of press. I believe that partial answers to many of the questions about SARS-CoV-2 can be found by pulling back, and looking the current viral outbreak from the perspective of where this virus fits into a broader group of viral relatives—its family. How have other members of this family evolved? What conditions shape the direction that natural selection takes and what might that tell us about the future of this virus and the potential for additional outbreaks?
What is coronavirus?
I’m not talking specifically about the strain of virus that is causing havoc right now across the world. Rather I’m interested in the relationships of this virus to other viruses that are collectively called coronavirus.
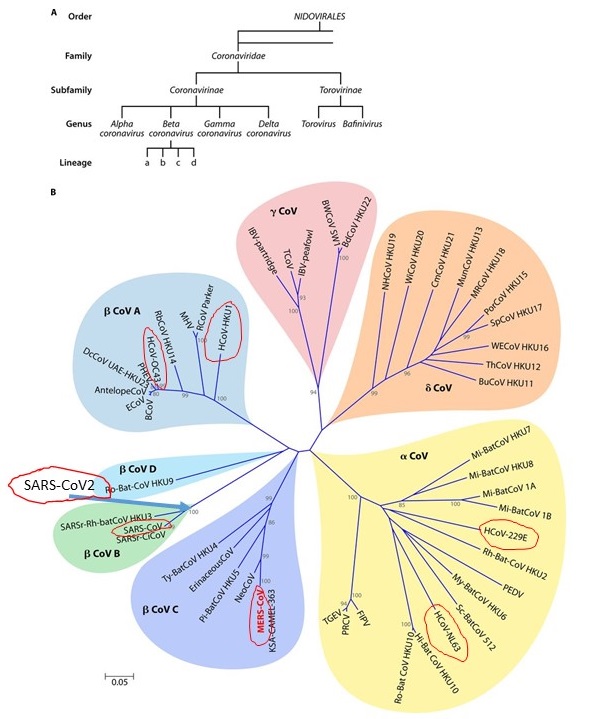
The term coronavirus refers to viruses that are classified as a family of enveloped, positive-sense, single-stranded RNA viruses. Usually the term refers to all the members of groups (genera) of viral “species” (see footnote 1 regarding species vs strain) referred to as the alpha, beta, delta and gamma coronavirus (Figure 1 below). There is a closely related group of viruses sometimes placed into—or sister to—this coronavirus family called torovirus which causes gastroenteric disease in cows, horses and pigs and that infects humans, usually children, as well (Koopmans 1997).
Coronaviruses infect a diverse set of animals including many species of birds, humans, bats, pigs, giraffes, beluga whales, bottlenose dolphins, hedgehogs, cows, antelope, camels, alpacas, dogs, cats, civets, pangolins, and likely hundreds of additional hosts that we have yet to discover. As we will see, the coronavirus family seems to be rapidly expanding its range as they continually invade new host species.
Bats and birds are the most frequently identified hosts infected by coronavirus. Bats are the most speciose of all major groups of mammals with 1200 species worldwide. Samples taken from bat species have detected hundreds of different strains of coronaviruses in all parts of the world. This is also likely true of many species of fowl and other bird forms.
Typically, a strain of coronavirus is unique—and therefore presumably adapted to—an individual species of bat or bird. However, the similarity of genome structure and RNA sequences among all coronavirus strains—or species if you prefer—strongly suggest that all bat, bird and coronaviruses inhabiting other mammals can trace their origins back to a single common ancestor coronavirus in the not too distant past.
That ancestral coronavirus likely had either a bird or a bat host. Subsequently, that ancestral lineage of coronavirus reproduced in its host some mutations provided a way of escape from that host species allowing it to invade another host species, and eventually adapting itself to that host species’ environment via additional mutations and natural selection acting on those mutations. From bats and birds, the virus has inexorably spread itself among a large number of mammalian and avian hosts.
Overview of the broader group which contains coronaviruses:
The coronavirus family is not completely unique among viruses. It is a members of a larger group of viruses (Order Nidovirales, see figure below) classified together because they share a common means of storing their hereditary material. They are positive-sense single-stranded RNA viruses. They store their genetic material on a single strand of RNA unlike all cellular-based living things which store their hereditary information in the form of double-stranded DNA. Many members of the Nidovirales cause disease in mammals and are usually enteric attackers (meaning they go through the stomach and attack the walls of the intestine causing dysentery and sickness in animals). Many members of the coronavirus attack the intestines as well, including the SARS-CoV-2 which has been reported to cause such symptoms in some individuals.
There are seven known species/strains of coronavirus that infect humans
Seven species human coronaviruses (HCoV) have been identified (Figure 1 and 2). Four of these produce generally mild symptoms typical of a common cold. Two of these (HCoV-229E and HCoV-NL63) are in the Alpha coronavirus group while the other two (HCoV-OC43 and HCoV-HKU1) are members of the Beta coronavirus group. All these viral species are circulating worldwide and cause symptoms in children and adults.
The other three species of human coronavirus that have entered into the human population cause much more severe symptoms and are all derived from the Beta coronavirus group. These are the now more familiar: Middle East respiratory syndrome-related coronavirus (MERS-CoV), Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), and Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2).
Of these SARS-CoV no longer is circulating among humans. It is known to have infected 8096 individuals causing 810 death between 2002 and 2004 before being completely contained. The MERS coronavirus (2012-present) has infected even fewer individuals (2586 known cases to date) and is communicated to humans from infected camels, who themselves contracted the virus from bats. In humans it causes severe symptoms and kills its human host 36% of the time (816 deaths to date). However, the virus does not transfer easily from human to human and thus a large number of new cases of MERS are the result of zoonotic (from animal host) infection. Its spread among humans is severely limited and therefore not likely to become a native human-spreading virus any time soon.
We know what SARS-CoV-2 is doing right now, having infected at least two million people with over 125 thousand dead as of April 14, 2020.
Where did the three most recent human coronavirus species come from?
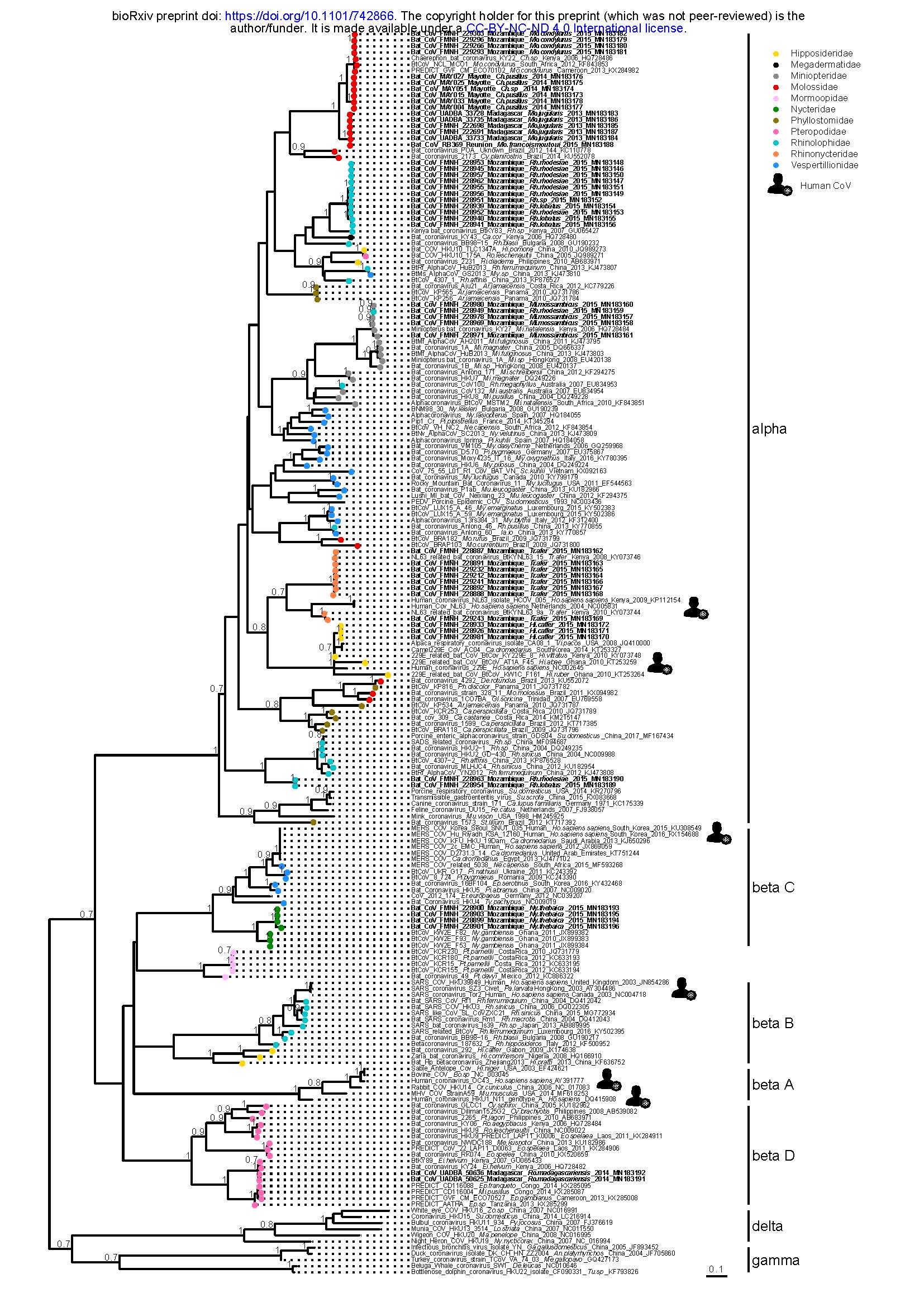
All species of coronavirus that infect humans, including the cold-causing species, are zoonotic in origin. This means they had another animal host before being transferred to humans. We can say with certainty that at least seven times in human history a coronavirus species from an animal has been able to make the jump from that animal to humans.
For example, as noted above, the MERS virus has been traced to a camel population, but it is likely that these camels are not the original host of the viral strain that has infected them and humans; rather its long-term host is likely a bat. Camels themselves are severely affected by the virus with high death rates suggesting that the virus has yet to adapt to the camel environment and become native to camels. DNA sequences of bat coronaviruses in species in the Middle East show very high similarity to the camel and human strains. MERS is a bit different from the other human coronavirus strains because it has yet to adapt to the human host system. Most cases of human infection are independent transfers from camels rather than human-to-human transfer. It appears that the virus has not yet achieved the right combination of changes to its genome to allow it to become a viable human-spread virus like the others human coronaviruses have.
What about the Beta coronavirus SARS strains that infect humans? The origin of SARS-CoV-2 is still debated but there is high sequence similarity of this virus to a virus in one bat species in china and coronavirus extracted from pangolins (Anderson et al 2020). Pangolins could have been an intermediate vector (bat to pangolin and then to human) or possibly it was bat virus to human and also the same bat virus to pangolin in a similar time frame. Regardless of the exact vector, unlike MERS which is independently transferred from camels to humans over and over again (rather than from camel to human and then human to humans), the SARS-CoV-2 virus probably was a one-time transfer that took hold in a human late last year (2019) and has been able to be transferred from human to human after that.
SARS-CoV in 2002/2003 was also a single transfer event but that time it was from a civet in southeastern Asia as an intermediate host (though the civet virus was likely derived from a bat species that had recently invaded the civet population).
What are the origins of other human coronavirus species?
What about those other cold-causing coronavirus strains? Interestingly, some of them are quite young as well. We don’t have specific reports of the first appearance of these other viral species probably because they did not cause symptoms that were different enough from other viral or bacterial diseases that we could recognize them at the time of their introduction into humans. However, we have sequenced their genomes and compared them to other viral species in other animals and through phylogenetic analyses and molecular dating methods we can estimate when each species shared a common ancestor with another animal version of the virus.
Those analyses suggest that all human coronavirus species are quite young: only a few hundred years old at most. HCoV-NL63 may have entered into humans as long ago as 800 years (Abdool Rasool et al 2010) but HCoV-OC43 finds its origins in the late 1800s and likely was derived from a cow coronavirus which also spread to horses and is a global problem for both of them today (Vijgen et al 2005). The others two originated in the 1900s and have genomes most similar to some bat coronavirus species. Human coronavirus 229E is most similar to known coronavirus from bats possibly with an intermediate through camels or alpacas (Figure 2 and Figure 3) and may not have entered into humans until the 1950s (Corman 2015).
It’s a numbers game
The prevailing idea is that bats and birds are the long-term hosts of many coronavirus species (there are more than 1000 bat species worldwide and many and maybe most harbor a unique species of coronavirus) and that from bats many other animals are infected with coronavirus. In those secondary hosts viral mutations are selection to better adapt that virus to its new hosts Many of those changes may also conveniently—for the virus—also made them more amenable to entry into human beings. Not all HCoV strains have the same means of entering into human cells. SARS-CoV finds its way in through our ACE2 cell-surface protein but other strains are adapted to different entry points on the cell surface. This may explain some of the differences in the progression and outcome of the disease caused by each HCoV strain.
As with all viruses, trillions of members of each viral strain are experiencing mutations at any moment in time, each of which has a chance of producing the right combination to find an entry into human beings. If a person happens to be in contact with that particular virus (eating that animal, near its feces, in its airspace when it coughs) at that moment that virus may infect a person and then there is a chance it will move from that person potentially beginning a new strain of virus that circulates among humans. No doubt, uncountable numbers of viruses have experienced random mutations that have provided them with the special combination to enter into humans but at the moment of that creation no human being was there to be infected and that particular mutant virus went extinct.
So it’s a numbers game. With so many opportunities cross-species infection is bound to happen and we can see that at least seven times in our history the right combination of mutations has occurred at a moment when there was a human being making contact with an infected animal.
Could there be more than seven origins of human coronaviruses?
Absolutely! We can be almost certain there have been many more introductions into humans. But we also know that those introductions were not completely successful or they would be endemic to humans today. Just five species of coronavirus are widely circulating in the human population today (MERS and SARS-CoV are not widely circulating). But that doesn’t mean that there haven’t been dozens if not hundreds of even thousands of times that a coronavirus has infected a single or a few human beings. Those introductions could have occurred anywhere humans come into contact with infected animals which would be just about anywhere on earth.
Many of these jumps to humans only infected a single person and the virus either wasn’t able to jump from human to human or that person died or quickly recovered before they could spread it to another person. Either the unique version of the virus died with its host or was simply killed off by that person’s immune system. Some of these may have resulted in pneumonia but since pneumonia is so common and caused by so many different diseases rarely are the infectious agents determined and so novel viruses are often overlooked. The fate of that particular origin in history was a quick extinction that we would never know about. We don’t know how many times this has occurred or is occurring today but we can be confident that it is happening—and with increased regularity.
What we do know is that population and evolutionary biology can tell us that the odds of successful transfer to human being has increased over time. We are more susceptible to novel coronavirus infections than we ever have been.
Why? There are at least two reasons: 1) the population density and mobility of human beings has increased over time and 2) the coronavirus family has grown more diverse and common over time. Let me explain why both of these factors put us at every increasing risk of more pandemic coronavirus infections in the future.
The population density effect
The easiest way to understand this is to ask yourself the question: were there coronavirus pandemics among humans thousands of years ago? The answer is – not likely. Why? Because if you were alive 5000 years ago and someone in your village contracted a coronavirus from an animal what would have happened? If the virus was able to pass from human to human it would have spread to everyone in the village. Maybe 20% of the villagers died or none of them died but once everyone had the virus the virus would run out of hosts and that particular viral strain would go extinct. This could have happened hundreds of thousands of time not just for cononavirus but any number of other viruses that are found in other animals with larger populations.
For any virus to persist it must find a continual string of new hosts. If that village didn’t have contact with others outside for two weeks the virus would be doomed and even if one person walked to another village that village would have to have occupants who travel within two weeks to another to keep it alive. Any successful virus must find a host population large enough that there will be constant contacts with new hosts and therefore continuous spreading opportunities.
Another strategy that aids in viral survival is its capacity to mutate over time. In a very large host population it is possible that a virus could eventually infect every individual but because it may take years for that to occur, by the time it has infected everyone the virus may have changed just enough that the immunity that individuals had gained will either be ineffective or will have been lost allowing the virus to re-infect the same individuals again. This is how common cold and flu viruses continue to persist over hundreds and thousands of years.
As we have become more densely populated and travel has become more common, it has opened us up as better viral vectors than we were in the past. When humans were living in small communities with infrequent contact, there were few viruses (there are exceptions but those viruses have different life cycles than the flu, cold, small pox, measles etc..) that could gain a strong foothold in the human population.
This general rule applies to most—though not all—viruses. Our ancestors simply didn’t have to deal with the same deadly viruses because when our populations were small and dispersed over large areas most viruses had no chance of surviving long enough in a human host to adapt to us before going extinct. Today, the human population is so large, dense and mobile that we form a very attractive host for many viruses.
Coronavirus expansion among mammal species has created more opportunities for host jumping
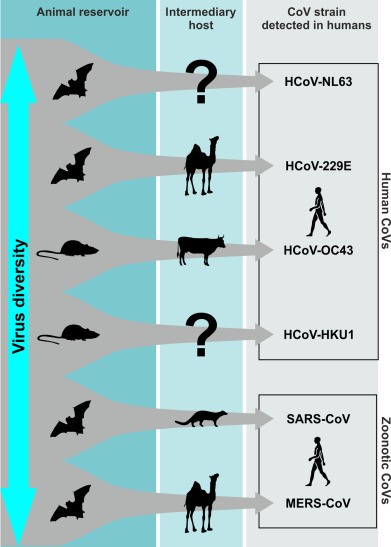
As I mentioned before, the coronavirus family is relatively young. It also is a highly successful group of viruses that is currently undergoing rapid expansion across the world. You can think of the family itself as constantly growing and spreading. It is invading new hosts and as it successfully adapts to those hosts, those hosts then create new opportunities for finding new hosts. Scientists exploring the evolution of this family of viruses think that all of the members of the family including those of the Alpha, Beta, Delta and Gamma Genera of coronaviruses all have a single common ancestor that lived possibly just tens of thousands of years ago. Compared to other viral families or families of other animals their genomes are amazingly youthful. The family itself seems to be closely related to another group of viruses called the toroviruses that have a similar genome structure and cause gastroenteritis infections as do many coronaviruses. They infect pigs, cows and horses and have been known to cross over and infect individual humans and children in particular. So if we traveled even further back in time we would likely discover that the toroviruses and coronaviruses also share a common ancestor from whom these now relatively distinct families evolved through changes as they invaded and adapted to new hosts and selection regimes.
The point is that biologist can use genome analyses and comparison of overall divergences of their RNA sequence to reveal a strong signal pointing to a recent common ancestor of all coronaviruses. That common ancestor would have been restricted to a single host species when it existed. That host appears to have been either a bat or a bird (birds possibly because two of the groups—delta and gamma—of the coronaviruses infect primarily birds today) from which the virus has subsequently spread into dozens of groups of animals and thousands of species over just a few thousand years. So if you were to go back in time, you would encounter fewer and fewer coronavirus species in fewer and fewer species of other animals. As a result the risk of contact and transfer from animal to human must also have been far less the further back in time you go.
Let me provide a few examples to illustrate how young many of these coronavirus species are. Scientists recently found coronavirus in a bottlenose dolphin (Woo et al 2014). When they examined its sequence they discovered that it was very similar to a coronavirus from beluga whale. They estimated that the virus in the two cetacean species shared a common ancestor as recently as 1959. And the beluga whale virus is not that dissimilar to a variety of Gamma coronaviruses that infect various bird species so this virus may have only invaded the cetacean family possibly directly from birds within the past several hundred years.
Likewise, the coronavirus which is pandemic among chickens around the world (does millions of dollars of damage to the poultry business) may have only originated a few hundred years ago (Cavanagh 2007). As a side note, scientists have developed a vaccine for the poultry version of the coronavirus. Unfortunately it doesn’t seem likely it will have any effect on the current SARS-CoV-2 virus even though they are in the same family of viruses.
Cow coronavirus seems to have started as a bat virus that jumped to cows. In cows that virus further mutated and was able infect horses and one of the strains of human coronavirus (HCoV-OC43) is and very close relative of the cow coronavirus. This strain is particularly common in the human population and it is likely that you have suffered from cold-like symptoms as a result of this virus as some point during your life.
You can think of the expansion of the family of viruses over time just like the expansion of a virus in a single host species. In order to survive it must find a new host. In a single species with high density and large population it can continue to persist, but if some viral particles can escape that host and enter into another, that is another way to persist over time. Coronavirus has found great success in bats and some birds as their probable original hosts but they have been expanding to new hosts which allow for new opportunities for even greater success measured as total number and diversity of the family. Many viruses in other families only rarely move from one host type to another (and have long histories with those hosts as a measure of their success) but coronaviruses have proven particularly adept at the strategy of host jumping as a way to expand quickly and take advantage of the diversity of life on earth.
Success of the virus in other animals combined with large human population size is a problem
Combining a rapidly evolving and expanding group of viruses over the past few thousand years along with a rapidly growing and increasingly dense and mobile populations of humans over the same time, we can infer that the risk of a coronavirus successfully colonizing human beings has gone from near zero in the past to high enough that it has now happened multiple times in the past 200 years. The odds of future intrusions into humans will continue to increase as more and more species of animals around us become potential carriers of coronavirus.
It’s not a question of if, but rather when, the next novel coronavirus outbreak will occur in humans. Virologists have been telling us this for the last two decades which is why research into the biology of this family has been especially important.
Natural selection and coronavirus
Why is this family of viruses so successful? Part of the answer may lie in the features of its life cycle. For the most part it causes mild symptoms without killing the host, and this allows the virus to spread readily. Coronavirus species may spread through particles in cough, feces, and can survive outside of the host cell for many days in or on certain substrates. This is a powerful combination of features.
A predication of viral evolutionary theory is that the longer a virus has persisted in a host species the more adapted it will become to that species. This is the result of natural selection at work. Variation resulting from mutations to its genome which promote features like stability outside the host and thus increase ease of transfer are selected (survive to the next generation) thus producing a new generation of viruses that are even better adapted to spread. Viruses that cause mild symptoms and yet high viral load allow the host to function alongside other hosts, thus increasing the opportunities to spread. In addition, mutations along the way along with the non-random sorting of those mutation by natural selection allow further adaption of the virus to its host ecology.
Overtime that “adaption” to the host may result in the progressive reduction of the damage done to the host species. Initially the virus may cause severe damage to the host species as it tries to copy itself as much as possible and is “maladapted” to its host. However, if the virus kills its host before passing itself to another host then the instructions in its genome that were so deadly to the host will not be passed to the next generation (we would say that that strain of virus had negative fitness or was under negative selection). On the other hand if there are mutations in the virus that cause it to slow the viral attack and allow the viral host to survive and pass the virus to another host—in the extreme this might be asymptomatic and yet contagious individuals—then the genetic combination that allowed for the virus to not be as aggressive in the host will survive and future generations of that virus may be less deadly. Over many generations of this form of selection, viral strains that were once very lethal will evolve through this process to become less lethal.
Social distancing and the direction of natural selection
Let’s apply this principle of natural selection to social distancing. Social distancing is an environmental effect and one that the virus must adapt to if it is going to survive. If a virus is highly lethal such as Ebola it tends to create natural social distancing which then leads to a reduction of vectors for the virus to continue its existence. As a result natural selection on a virus experiencing reduced host contact will select for viral strains that are less lethal and probably more importantly strains of virus that have a longer life cycle in the host. The latter will counteract the social distancing. If the virus can live in the host for 14 or more days and be infective during that time but not cause severe symptoms there is a good chance the virus will find another host. In the case of Ebola the lack infections allowing for greater opportunities for mutations and natural selection has not allowed it to adapt to the human ecosystem. It is just too deadly to become widespread and thus create the conditions for its own evolution.
For SARS-CoV-2, it fortuitously—from the virus perspective—arrived on the scene amazingly adapted to overcome our distancing measures. It can persist in teenagers for long periods of time without causing noticeable or strong symptoms. As a result it is hard for those harboring the virus—especially if they don’t know they have it—to distance themselves long enough for their bodies to kill the virus. They spread the virus unknowingly thereby passing strains of the virus that are well adapted to more and more hosts.
A general expectation is that more distancing should result in selective pressure on the virus to preferentially propagate viral forms that are less lethal over time. On the other hand, a large population living close together can have the opposite effect. If strains of the virus cause severe outcomes but are still able to spread there is selection that happens within the individual host for viruses that propagate faster and thus can do more damage.
Imagine a cell that was infected by a coronavirus goes on to make 10,000 new viral particles. As it makes those new particles many will have unique new mutations (eventually a person with a COVID-19 infection may generate several trillion viruses! – see Footnote 2). When they spill out into the respiratory track and look for other cells to invade, the ones with mutations that allow them to get into new host cells faster will outcompete the ones with mutations that may have slowed the process of infection. The ones that infected cells faster will have a greater opportunity to make even more viruses, many of which will have the same properties and thus create an even higher percentage of viruses that infect even faster. This can cause the virus propagating in a single person to increase its efficiency and thus may cause more severe symptoms. If there is a large population living next to each other those severe cases will still result in spread. So some of the viruses will escape to a new host and continue to cause severe symptoms in them as well. As long as there are enough victims to continue the viral lineage, selection for more aggressive strains may be greater than any selection for milder strains and therefore push the population of viruses toward viral strains that are more virulent over time.
Of course selection in the direction of higher virulence can only go on for so long. At some point the virus will run out of hosts or become so virulent that people will naturally start to physical distance such as happens with Ebola virus. And when that happens the selective pressure pushes harder the other direction—toward viral strains that are less virulent because they are more likely to survive if they don’t cause strong symptoms.
Some might wonder, what about those “mild” strains of human coronavirus? Might they have started out initially with more pronounced effects on humans? Yes, it is quite possible that the other HCoVs may have caused more pronounced disease upon first jumping to humans. There are a few people that hypothesized that the 1890 pandemic might have been related to the origin of the now common HCoV-OC43 strain which has subsequently become less virulent over time (Vijgen et al 2005). More detective work seems to have dismissed this hypothesis in favor of a traditional influenza virus but the fact that this was proposed illustrates that it is not uncommon to understand the many viruses are far more damaging when they first jump species and then are shaped by natural selection into milder forms. We see the same thing in many strains of HIV after it jumped from primates to humans.
Natural selection is a complicated phenomenon that takes into account hundreds of environmental variables. But it invariably shapes the pattern of adaption of all living things within their environment. Understanding which of variables are the strongest factors in natural selection is crucial for predicting how a virus will adapt. The more we study the variables involved and measure the strength of each variable in the selection process the better we can predict the path viral evolution will take. Even the development of a vaccine for a novel virus involves a large measure of understanding the evolution of the virus and its genome. We have to anticipate what changes the virus may experience as a reaction to the selective landscape in the human body.
Why don’t coronavirus species cause death rates of 1 to 10% in other animals as they have in humans?
First, coronavirus do cause disease and even deaths in many species of animals. The scourge of coronavirus on poultry and cattle is evidence of that. It is much more difficult to quantify the extent of illness that the virus may cause among wild populations but it surely lowers fitness of many individuals.
It very well may be that coronaviruses have killed several percent of natural populations but it’s unlikely that we would notice if 1 or 2 percent of a particular bat or duck or mouse population were to succumb to a new viral strain over a several month period. In wild populations the result of such a strong selection event would be a surviving population that would be more resistant to the deadly effects of the virus. Today, we observe coronavirus circulating at high frequencies in some animal species with no obvious large effects. This is usually an indicator that that species has lived with that virus circulating amongst its members for decades to thousands of years.
Obviously human beings are aware of what happens to other members of our population in a way that we are not aware for other animals. We can recognize when deaths are occurring at a rate higher then natural background levels and then respond.
A rather difficult way to think about our current predicament with SARS-CoV-2 is to realize that had this virus entered into the human race 1000 years ago we might not have noticed. Not because we just didn’t have the sensitivity in our health system back then to realize what was going on, but in all likelihood the death rate could have been much lower in the past than in the present despite our better health system.
Why? Painting with a very broad brush, because in the past a much smaller portion of the population would have been 65 years or older. Those that were 60 would have been less likely to be obese, have heart disease or have compromised immune systems due to cancer treatments and so forth. In other words, in a crude natural selective sense, the majority of those dying from coronavirus today would have already been dead of other causes in the past. This in no way justifies not treating individuals for these conditions which we can do and establish a good quality of life far beyond what was possible in the past. But it is a distinguishing characteristic of current outbreaks of both the flu and other viruses in our modern times. In populations of wild animals the pool of compromised individuals is far lower—by percentage—and so we should not be surprised to see the same virus having lower fatality rates in those populations.
Nevertheless, it is not possible for us to predict a priori how any particular species will be affected by a particular virus in its onset. The fact we have seven different coronavirus strains that have infected humans each of which cause different forms of disease is evidence of the varied responses that viruses can elicit.
SARS-CoV-2 – Natural or Lab made?
A large fraction of the American public seems to believe the virus circulating today was either made in a lab or the result of an accident in a lab. Is this possible or reasonable?
Possible as an accident? Yes. Likely? No. The result of bioengineering? Almost certainly not. Numerous scientists have examined the genome structure of the new viral strain and concluded that the virus is naturally formed (Anderson et al 2020, Tang et al 2020) even if that happened in a lab studying coronavirus. The fact that very similar strains of virus are in natural populations of bats, pangolins and possibly canines (Xia 2020) and the knowledge that natural transfers from animals to humans have been successful at least six times in the past should place the burden of proof on those that make these claims.
What is most important is that genome sequencing points strongly to a single origin of all SARS-CoV-2 strains of this virus now circulating among humans rather than multiple origins like in MERS. This probably means this was a mutation in a single individual animal or small population and it is unlikely that this same viral strain is out there waiting to come right back into the human population though similar strains may yeild very similar SARS-like jumps again. The MERS strain which exists in a reservoir of camels and bats continues to come back over and over again.
The future of SARS-CoV-2?
At this point it is evident that this virus has become the 5th endemic coronavirus of human beings and will circulate among humans for many generations to come. As it does so it continues to evolve through mutations (Figure 4 below) and natural selection. What this virus looks like in a few years is what everyone wants to know. I don’t know but I would imagine it will become something like the flu. It will be a virus that may lose some of its punch over time as humans become more resistant. That will slow its spread and spread out the load on health care but it will likely continue to be a serious concern for the elderly just as many strains of the flu are. In a year or two many of us will be going to get a flu and a coronavirus vaccination. At that time it will be manageable and we will simply be waiting around for the next coronavirus to pop into the human population. Maybe it will just be a cold or maybe it will be more like the first SARS. Hopefully we will be better prepared. We will know more about the family and we will have a head start on vaccine production.
Could human coronavirus escape into other species?
Yes! Patrono (2016) reported direct evidence that chimpanzees had been infected by human HCoV-OC43 strain and caused cold-like symptoms. The ACE2 receptor that the SARS-CoV-2 virus uses to gain entry is identical to humans in protein structure at the point of viral attachment in the great apes and other primates suggesting that this new virus could be transferred from us to other primates. Corman et al (2018) brings up an interesting observation regarding primate coronaviruses. They observe that coronaviruses are at best rare in chimpanzees and other apes despite the fact that they should be as susceptible as human are to catching these viruses. They suggest that the lack of coronavirus in apes may lend support to the idea that most human coronaviruses are the result of humans having large amount of contact with domesticated animals or wild meat markets. Other primates would not have such contact and so have fewer opportunities for coronavirus transfers. Many domesticated animals (cows, dogs, chickens) live in high density populations which are natural breeding grounds for viruses and we spend a good amount of time with these animals.
Many of you may have also heard that tigers from the Bronx Zoo tested positive for COVID-19. Molecular biologist believe that the receptors on felines may be more susceptible to this new virus than canines so it is possible that we may infect cats and form a new coronavirus strain in them. Domesticated cats already have another widely shared strain of coronavirus (Addie and Jarret 1992) so again we have history to tell us it is possible. We don’t know the consequences to these animals is that jump takes place. The virus could have a worse or better outcome in other species but we do know that this is what coronaviruses do—they extend their reach into other species over time as they continue to advance their spread through the animal kingdom.
Final thoughts
How this latest incursion of coronavirus into humans will eventually play out is a story yet to be written. But we can say that little of what has happened is a surprise. Coronaviruses were targeted as a group that had potential for causing pandemics even before the SARS outbreak in 2002. The “mild” strains are all known to cause severe acute respiratory syndrome in some patient and it has been apparent that many of these strains were only recently introduced into humans so the possibility of the origin of additional stains should be of no surprise to anyone. This is especially true after 2002 and even more so after 2012.
The use of phylogenetic analyses, genome sequencing, to inform and understanding how the evolutionary mechanisms at play in the origins and continuing development of this emerging family of viruses have played a key role in helping us to predict what we are witnessing today. For my class I just recorded a lecture on the evolution of pathogens examining the flu, the natural selection dynamics of antibiotic use, cancer formation and treatment, and the origin of new diseases. It’s something of a capstone experience pulling together a variety of themes from the course and applying it to how organisms adapt and change to their environment. Understanding these principles plays a critical role in their ability, many as future doctors, to synthesize and react to new diseases, help inform policy decisions and communicate with the public.
Footnotes:
- I use the terms “strain” and “species” interchangeably with regard to viruses that in various hosts. These are both used in the literature. It is hard to relate viral “species” to species of other organisms but the idea is that a lineages of viruses that have a common ancestor and inhabit the same host species is a good approximation of a viral “species” as well. But the language of strain is also appropriate just as microbiologists refer to strains of bacteria rather than species or at times a stain can be thought of a subspecies. Either way, the idea is that a strain or species are a group of related (using by RNA sequence) viruses that share a relatively recent common ancestor.
- Viral population numbers always astonish me. I mentioned that a single individual with a coronavirus or flu virus infection may harbor 2 trillion viruses at the peak of their infection. Some have estimated that a cough from an infected person may spew out 200 million virus capsules. If you consider that a million people in the US have been infected with the coronavirus you could reasonably conclude that there some 2,000,000,000,000,000,000 viruses contained in those infected individuals right now. That is two quintillion!
Viruses are the most common life form—if you consider them alive—on the face of the earth. It is estimated that there at least 1 with 31 zeros behind it viruses in organisms right now. That is more than the number of stars in the Universe. Carl Zimmer has written a whole book on the world of viruses (A Planet of Viruses) but this interview from NPR is a nice summary of the ubiquity and important of viruses to the ecology of the Earth. https://www.npr.org/2011/05/06/136057352/carl-zimmer-explores-the-weird-lives-of-viruses
References
Abdul-Rasool, Sahar, and Burtram C. Fielding. “Understanding human coronavirus HCoV-NL63.” The open virology journal 4 (2010): 76.
Addie, D. D., and O. Jarrett. “A study of naturally occurring feline coronavirus infections in kittens.” The Veterinary Record 130, no. 7 (1992): 133-137.
Andersen, Kristian G., Andrew Rambaut, W. Ian Lipkin, Edward C. Holmes, and Robert F. Garry. “The proximal origin of SARS-CoV-2.” Nature Medicine (2020): 1-3.
Borucki, Monica K., Jonathan E. Allen, Haiyin Chen-Harris, Adam Zemla, Gilda Vanier, Shalini Mabery, Clinton Torres, Pamela Hullinger, and Tom Slezak. “The role of viral population diversity in adaptation of bovine coronavirus to new host environments.” PloS one 8, no. 1 (2013).
Cavanagh, D (2007). “Coronavirus avian infectious bronchitis virus”. Veterinary Research. 38 (2): 281–97. doi:10.1051/vetres:2006055. PMID 17296157.open access
Chan, Jasper FW, Susanna KP Lau, Kelvin KW To, Vincent CC Cheng, Patrick CY Woo, and Kwok-Yung Yuen. “Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease.” Clinical microbiology reviews 28, no. 2 (2015): 465-522.
Corman, Victor Max, Heather J. Baldwin, Adriana Fumie Tateno, Rodrigo Melim Zerbinati, Augustina Annan, Michael Owusu, Evans Ewald Nkrumah et al. “Evidence for an ancestral association of human coronavirus 229E with bats.” Journal of virology 89, no. 23 (2015): 11858-11870.
Corman, Victor M., Doreen Muth, Daniela Niemeyer, and Christian Drosten. “Hosts and sources of endemic human coronaviruses.” In Advances in virus research, vol. 100, pp. 163-188. Academic Press, 2018.
Holmes, Edward C., and Andrew Rambaut. “Viral evolution and the emergence of SARS coronavirus.” Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 359, no. 1447 (2004): 1059-1065.
Joffrin, Léa, Steven M. Goodman, David A. Wilkinson, Beza Ramasindrazana, Erwan Lagadec, Yann Gomard, Gildas Le Minter et al. “Bat coronavirus phylogeography in the western Indian Ocean.” bioRxiv (2019): 742866.
Koopmans, Marion PG, E. Simone M. Goosen, Aldo AM Lima, Isabel T. Mcauliffe, James P. Nataro, Leah J. Barrett, Roger I. Glass, and Richard L. Guerrant. “Association of torovirus with acute and persistent diarrhea in children.” The Pediatric infectious disease journal 16, no. 5 (1997): 504-507.
Patrono, Livia V., Liran Samuni, Victor M. Corman, Leila Nourifar, Caroline Röthemeier, Roman M. Wittig, Christian Drosten, Sébastien Calvignac-Spencer, and Fabian H. Leendertz. “Human coronavirus OC43 outbreak in wild chimpanzees, Côte d´ Ivoire, 2016.” Emerging microbes & infections 7, no. 1 (2018): 1-4.
Saif, Linda J. “Bovine respiratory coronavirus.” Veterinary Clinics: Food Animal Practice 26, no. 2 (2010): 349-364.
Tang, Xiaolu, Changcheng Wu, Xiang Li, Yuhe Song, Xinmin Yao, Xinkai Wu, Yuange Duan et al. “On the origin and continuing evolution of SARS-CoV-2.” National Science Review (2020).
Vijaykrishna, D., Gavin JD Smith, Jing Xua Zhang, J. S. M. Peiris, Hongling Chen, and Yi Guan. “Evolutionary insights into the ecology of coronaviruses.” Journal of virology 81, no. 8 (2007): 4012-4020.
Vijgen, Leen; Keyaerts, Els; Moës, Elien; Thoelen, Inge; Wollants, Elke; Lemey, Philippe; Vandamme, Anne-Mieke; Van Ranst, Marc (February 2005). “Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event”. Journal of Virology. 79 (3): 1595–1604. doi:10.1128/JVI.79.3.1595-1604.2005. ISSN 0022-538X. PMID 15650185.
Woo, Patrick CY, Susanna KP Lau, Carol SF Lam, Alan KL Tsang, Suk-Wai Hui, Rachel YY Fan, Paolo Martelli, and Kwok-Yung Yuen. “Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus.” Journal of virology 88, no. 2 (2014): 1318-1331.
Xuhua Xia, Extreme genomic CpG deficiency in SARS-CoV-2 and evasion of host antiviral defense, Molecular Biology and Evolution, , msaa094, https://doi.org/10.1093/molbev/msaa094

Next week a paper from my hand will appear in a science journal demonstrating that SARS-CoV2 is just a variant of SARS-CoV of 2003. And most likely it hid and reemerged from bats: The SARS virus strain in bats (RaTG13) is >96% identical to SARS-CoV2. Leaving out the indels (as evobiologists do when comparing human and chimp DNA) they are almost 99% identical.
LikeLike
Appreciate the info. Interesting. A number of quibbles, but i address one specifically. You may find yourself incredibly embarrassed when all is found out. It did come from the virology lab which experiments on different viruses. Numerous sources have confirmed this and you should have noticed the last few days reports coming out to this effect (unless of course, you only watch CNN et.al. for your news). Both Chinese and American researches were working together as recently as last year in the lab at UNC and later at Wuhan. This virus is not JUST a naturally occurring virus. It has been altered (in the labs?). It did not come from the wet market (no bats were being sold there at the time). Our military reported as long ago as 2018 that the Wuhan lab had very poor preventative measures in place (in other words, it wasn’t if but when this would happen). Thus the cover up by the Chinese govt., the silencing (even jailing) of their scientists who were warning the scientific community, the destruction of lab samples and study papers, the removal of foreign journalists and investigators, etc.
Probably the virus is a result of military experiments with corona viruses (you can guess for what purposes) which infected workers there and then to the wet market, and sadly, to the rest of the world. Scientists playing god (or maybe just trying to develop vaccines) and carelessly allowing the infection and spread with lax security procedures. Evolution and natural selection had NOTHING to do with this.
LikeLike
Thanks, Joel, for doing this. Have you considered blogging on the status of testing for SARS-CoV2? That seems like the greatest stumbling block to reopening the country for business. What is holding up our ability to provide reliable, safe, and quick testing methods, for those who do not show outward symptoms of infection?
It seems like a lot of Christians are buying into the idea that the virus was introduced by this lab in Wuhan. Some go so far as saying that this was intentional, by the Chinese government, which is just crazy conspiracy theory. But a lab accident? Well, this is difficult to rule out completely, but you have made a good case to say that the burden of proof is on those who insist that this was derived in a lab, and subsequently leaked out into the public, due to some sort of carelessness.
Even the esteemed apologist William Lane Craig is drawn to the lab accident hypothesis, which I frankly found shocking:
LikeLike
While certainly not proof, this is the most complete collection of the circumstantial evidence for a lab accident I have found:
https://www.nationalreview.com/2020/04/coronavirus-china-trail-leading-back-to-wuhan-labs/
I agree completely that the intentional release is pure conspiracy delusion (if true, why would China have released the gene sequence to the rest of the world so quickly? That being just one of many problems with the idea).
LikeLike
Incredible to me that after a long post of well-reasoned, clearly-explained science 3 of 4 comments here are about conspiracy theories. Joel’s next post is gonna have to be about the crazy-conspiracy-viruses that infect people’s thinking. Fortunately, Joel is good at those kind of posts.
LikeLike
zach, something being well reasoned does not equate to it being true. And it is not just christians who think this originated in the lab? Do you just see it as a coincidence that a virology lab is a few hundred feet from the market. That chinese and american researchers have been working together both in wuhan and at unc on corona viruses. That the Chinese destroyed original samples and papers on covid-19? Why do this if it was just a naturally occuring virus from batman’s cave. I think you both have fallen into the “darwin of the gaps” fallacy where evolution can explain everything.
LikeLike
Joel is putting together facts based on scientific inquiry that leads to clarity about the truth.
You are putting together coincidences posed as rhetorical questions to make them sound convincing.
The first is worth listening to because it deals with evidence.
The second is folly because it’s conspiracy theory.
LikeLike
well zach if wishes were horses………. No, just reporting what anyone who wants to take the time and make the effort can find out for themselves, instead of swallowing wholesale a priori darwinian idealism and if you really believe that the darwinian paradigm is made up only of bunches of facts and truths, perhaps you’ve never actually read it through. There are few places where one can find more hyperbole and speculation, and non sequitors, than a darwinian evolution site. Of course you offered no refutation of my points, just the usual smug dismissals from the deluded.
LikeLike